November 2020 Case
Authors
Aidas J. Mattis, MD, PhD (Fellow) Jean Hou, MD (Faculty)
Renal Pathology
Clinical History
A Hispanic female in her 50s was referred to a nephrologist by her primary care provider for mild proteinuria with worsening chronic kidney disease. She has past medical history of hypertension, obesity and chronic kidney disease. Her kidney disease has slowly progressed over many years with her baseline serum creatinine now ranging from 1.7-2.0 mg/dL.
The patient has no history of diabetes. She takes lisinopril for her hypertension and no other medications on a regular basis. She denies NSAIDs use.
The patient has no known family history of kidney disease. The patient reports no known drug allergies. She does not smoke, drink alcohol, or use any illicit drugs.
On physical exam she appears well and her stated age. Vitals are unremarkable and the remainder of the physical exam is unremarkable.
Laboratory studies are notable for CBC and chemistry with all parameters within normal limits, Urinalysis is remarkable for 1+ proteinuria and 0+ blood. Serum creatinine is 2.0 mg/dL. Random urine protein is present, but reported as less than 200 mg.
A renal biopsy is performed.
Renal Biopsy
The portion of the specimen for light microscopy shows 18 glomeruli, 5 of which are globally sclerotic. The remaining glomeruli show extensive mesangial infiltration by amorphous, eosinophilic, and acellular material that stains negatively with PAS and silver stains, but blue-gray by trichome. (Figure 1A and 1B). The amorphous material is also extensively deposited in the cortical interstitium and some of the arterial walls (Figure 1A). A Congo red stain is performed, and stains positively within the amorphous material, exhibiting apple-green birefringence when viewed with polarized optics, consistent with amyloidosis (Figure 1C and 1D). Immunohistochemical staining revealed strong positivity for Leukocyte chemotactic factor 2 (LECT2) (Figure 1E). Proximal tubules show acute injury and necrosis with cytoplasmic protein reabsorption droplets. No atypical or fractured casts were present. Tubular atrophy involves approximately 50% of the tubules. Arteries show mild intimal fibrosis and muscular hypertrophy.
Immunofluorescence, on a scale of 0-4+ shows segmental amorphous staining for IgM (3+), C1q (3+), and C3 (4+) within the insudates. All other immune reactants are negative in glomeruli and within the amorphous deposits.
Electron microscopy reveals glomerular, interstitial, and vascular infiltration by non-branching, randomly oriented fibrils of indeterminate length and measuring approximately 10 nm in diameter (Figure 1F). In the glomeruli, the fibrillar material expands the mesangium and extends from the mesangial regions along the subendothelial aspect of adjacent glomerular basement membranes to produce segmental double contoured basement membranes. There are no tubuloreticular inclusions identified. Podocytes over non-infiltrated glomerular basement membranes show only mild podocyte foot process effacement, although there is extensive effacement over basement membranes heavily infiltrated by amyloid.
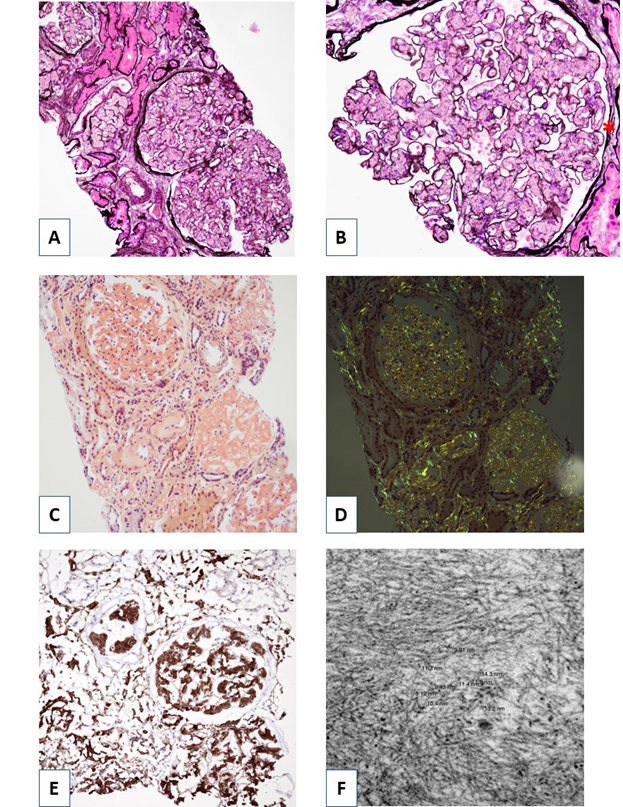
Figure 1. Renal biopsy findings. A) Jones methenamine silver stain, low power view demonstrating prominent glomerular mesangial infiltration by amorphous, eosinophilic, and acellular material. Silver negative deposits are also present in the interstitium. B) Higher power view of a single glomerulus highlighting mesangial infiltration by amorphous material. The amorphous material is focally associated with glomerular basement membrane duplication (asterisk). C) Congo red stain highlighting positively staining amorphous material in glomeruli, interstitium, and walls of some arterioles. D) Congo red stain, when viewed with polarized optics, reveals apple-green birefringence consistent with amyloid. E) Immunohistochemical staining for LECT2 is strongly positive within the Congo red positive material. Note the extensive cortical as well as glomerular infiltration. F) Electron microscopy reveals non-branching, randomly oriented fibrils of indeterminate length measuring approximately 10 nm in diameter.
Diagnosis
Amyloidosis, ALECT2 type; Acute tubular necrosis; Mild arteriosclerosis
Discussion
Here we present a case of a Hispanic female in her 50s who has had progressive chronic kidney disease with non-nephrotic range proteinuria. The renal biopsy showed extensive deposition of an amorphous, eosinophilic, and acellular material within the mesangial regions of glomeruli, the interstitium, and arterial walls. This material was confirmed to be amyloid by Congo red stain, with typical apple-green birefringence visible by polarization. Ultrastructural analysis further revealed typical non-branching and randomly oriented fibrils which measured 10 nm in diameter, on average. Immunohistochemical staining confirmed a diagnosis of leukocyte chemotactic factor 2 (ALECT2) amyloidosis.
Amyloidosis is the abnormal extracellular deposition of misfolded insoluble protein fibrils within tissues, predominantly as anti-parallel beta pleated sheets. Organs frequently affected by amyloidosis include the heart, liver, spleen, nervous systems, gastrointestinal, and kidneys (which are commonly involved). Diagnosis of amyloidosis commonly occurs in older males in the 6th or 7th decade of life, though it is seen in females frequently as well. There are now over 30 different types of amyloidosis that have been described in humans. The clinical outcome is dependent on the type of amyloid and the extent of organ involvement. The prevalence of amyloidosis varies geographically, with immunoglobulin light chain-derived amyloidosis (AL) most common in the developed world and secondary amyloidosis (AA) is most common in the developing world, most likely secondary to chronic infectious disease. More recently, ALECT2 amyloid was described in 2008 and has now become the third most common type of amyloidosis in kidneys and the second most common in certain ethnic populations [1-3].
Patients with renal amyloidosis (particularly in cases with extensive glomerular infiltration) frequently present with proteinuria which can range from minimal and asymptomatic to nephrotic range with full nephrotic syndrome. However, amyloid with vascular or interstitial-predominant deposition are more likely to be associated with progressive renal failure and less proteinuria. Diagnosis of renal amyloidosis requires a renal biopsy. Light microscopy often reveals variable deposition of amorphous, acellular and eosinophilic material which can occur in all renal compartments including glomeruli, interstitium, and vessels. The amorphous material stains weakly with periodic acid-Schiff (PAS) and negatively by Jones methenamine silver stain. On trichrome, the amyloid deposits often exhibit a blue-gray appearance. Confirmation that the amorphous deposits are amyloid occurs with positive Congo red staining and the presence of typical apple-green birefringence when viewed with polarized optics. Alternatively, staining with thioflavin T can show intense yellow-green fluorescence. Immunofluorescence studies are typically non-contributory for ALECT2 and AA amyloid, but can be diagnostic for AL amyloid if it demonstrates light-chain restricted staining for either kappa or lambda light chains. Electron microscopy demonstrates randomly-oriented, non-branching fibrils with an average diameter of 7-12 nm. In cases where immunofluorescence and immunohistochemistry are negative, the amyloid can be definitively subtyped by laser dissection and mass-spectrometry.
Different types of amyloidosis tend to deposit preferentially in different compartments of the kidney and where these deposits occur in each type of amyloidosis can have prognostic significance. AL can deposit in any renal compartment, including glomeruli, interstitium, vasculature and tubules and presents commonly with nephrotic syndrome with renal insufficiency. AA amyloidosis most commonly demonstrates predominantly glomerular and vascular deposition and present with proteinuria, nephrotic syndrome, and frequently renal insufficiency [2]. Glomerular deposits for AA patients tend to have poor prognosis with progression to end stage renal disease in 85% over a 5-year period [4]. ALECT2 amyloidosis presents with variable proteinuria which is absent in 1/3 of patients based on one study [5]. ALECT2 tends to have more significant deposition in the cortical interstitium with variable glomerular and vascular involvement [1, 2, 6]. ALECT2 patients have better overall survival than AL or AA patients, possibly because the cardiac tissue is rarely involved by amyloid, but ultimately up to 39% of patients will progress to end stage renal disease by one study [5].
ALECT2 amyloidosis was discovered by Benson et al in 2008, most commonly in Hispanic patients. Since then, ALECT2 has been reported in other ethnic groups including Chinese, Egyptians, First Nations People from Canada, Native Americans, Punjabi, and Sudanese in a series of case reports and larger studies [5, 7-13]. One of these studies looked at amyloid type among Egyptians and found that ALECT2 was the second most common type of renal amyloidosis, after AA, making up 31% of 116 cases. Pathogenesis of ALECT2 amyloidosis is not well understood. Although no mutations were identified in LECT2-encoding exons, it was noted that 10 out of 10 patients in one study were all homozygous for the G allele encoding valine at position 40 in the mature protein [6]. One study showed that there was recurrence in one patient of ALECT amyloidosis post-transplant, suggesting that ALECT2 is caused by circulating factors [5]. Median survival was 62 months with the best independent predictor of renal survival being serum creatinine of 2.0 mg/dL as the best cutoff for predicting end stage renal disease [5].
Treatments for AL amyloid include treatment of the underlying hematologic cause of monoclonal protein and/or plasma cell clone [14]. Treatment for AA amyloid involves treatment of the underlying inflammatory process which results in decreased production of the fibril precursor serum amyloid A protein, an acute phase reactant [15]. There currently is no specific therapy for ALECT2 amyloidosis.
Here we have discussed a brief review of the most common amyloid types and specifically focused on a relatively new amyloid type known as ALECT2 that is frequently identified in Hispanic and Egyptian populations. As the pathophysiology of ALECT amyloidosis is poorly understood and no treatments are available, further work is needed.
References
1. Benson MD, James S, Scott K, Liepnieks JJ, and Kluve-Beckerman. Leukocyte chemotactic factor 2: A novel renal amyloid protein. Kidney Int. 2008;74(2):218
2. Giannini G and Nast CC. An Organ System-Based Approach to Differential Diagnosis of Amyloid Type in Surgical Pathology. Arch Pathol Lab Med. 2020;144;3:379
3. Larsen CP, Beggs ML, Wilson JD, Lathrop SL. Prevalence and organ distribution of leukocyte chemotactic factor 2 amyloidosis (ALECT2) among decedents in New Mexico. Amyloid 2016;23:119
4. Uda H, Yokota A, Kobayashi K, Miyake T, Fushimi H, Maeda A, Saiki O. Two distinct clinical courses of renal involvement in rheumatoid patients with AA amyloidosis. J Rheumatol. 2006;33(8):1482
5. Said SM, Sethi S, Valerie AM, Chang A, Nast CC, Krahl L, Molloy P, Barry M, Fidler ME, Cornell LD, Leung N, Vrana JA, Theis JD, Dogan A, Nasr SH. Characterization and outcomes of renal leukocyte chemotactic factor 2-associated amyloidosis. Kidney Int. 2014; 86:370
6. Murphy CL, Wang S, Kestler D, Larsen C, Benson D, Weiss DT, Solomon A. Leukocyte chemotactic factor 2 (LECT2)-associated renal amyloidosis: a case series. Am J Kidney Dis. 2010;56(6):1100
7. Wang Y, Wang S, Zhang Y. Leukocyte chemotactic factor 2 associated renal amyloidosis: one case report. J Peking Univ 2015;47: 349
8. Larsen CP, Ismail W, Kurtin PJ, Vrana JA, Dasari S, and Nasr SH. Leukocyte chemotactic factor 2 amyloidosis (ALECT2) is a common form of renal amyloidosis among Egyptians. Modern Pathology (2016) 29, 416
9. Hutton HL, DeMarco ML, Magil AB et al. Renal leukocyte chemotactic factor 2 (LECT2) amyloidosis in first nations people in northern British Columbia, Canada: a report of 4 cases. Am J Kidney Dis 2014;64: 790
10. Larsen CP, Kossmann RJ, eggs ML et al. Clinical, morphologic, and genetic features of renal leukocyte chemotactic factor 2 amyloidosis. Kidney Int 2014; 86: 378
11. Dogan A, Theis JD, Vrana JA. Clinical and pathological phenotype of leukocyte cell-derived chemotaxin-2 (LECT2) amyloidosis (ALECT2). Amyloid 2010;17:69
12. Kulkarni U, Valson A, Korula A et al. Leukocyte derived chemotaxin 2 (ALECT2) Amyloidosis. Mediterr J Hematol Infect Dis 2015;7:32015043.
13. Holand DG, Acharya VK, Dogan A et al. Atypical presentation of atypical amyloid. Nephrol Dial Transplant 2011; 26:373
14. Leung N, Dispenzieri A, Lacy MQ, Kumar SK, Hayman SR, Fervenza FC, Cha SS, Gertz MA. Severity of baseline proteinuria predicts renal response in immunoglobulin light chain-associated amyloidosis after autologous stem cell transplantation. Clin J Am Soc Nephrol. 2007;2(3):440
15. Sack GH Jr. Serum amyloid A - a review. Mol Med. 2018;24(1):46