February 2020 Case
Authors
Andrew Siref, MD (Fellow), Serhan Alkan, MD (Faculty)
Subject: Hematopathology
Clinical History
Caucasian female in her 40’s with severe idiopathic aplastic anemia, on cyclosporine and promacta. Prior bone marrow aspiration and biopsy (performed at an outside institution) revealed a hypocellular marrow (20%) for age without dyspoiesis. She has periodically required platelet and red cell transfusions.
Laboratory Studies
CBC:
- WBC: 2.8
- Hb: 8.8
- MCV: 118.2
- Plt: 34
- Peripheral blood smear shows pancytopenia
LDH: 657 U/L
Flow cytometry
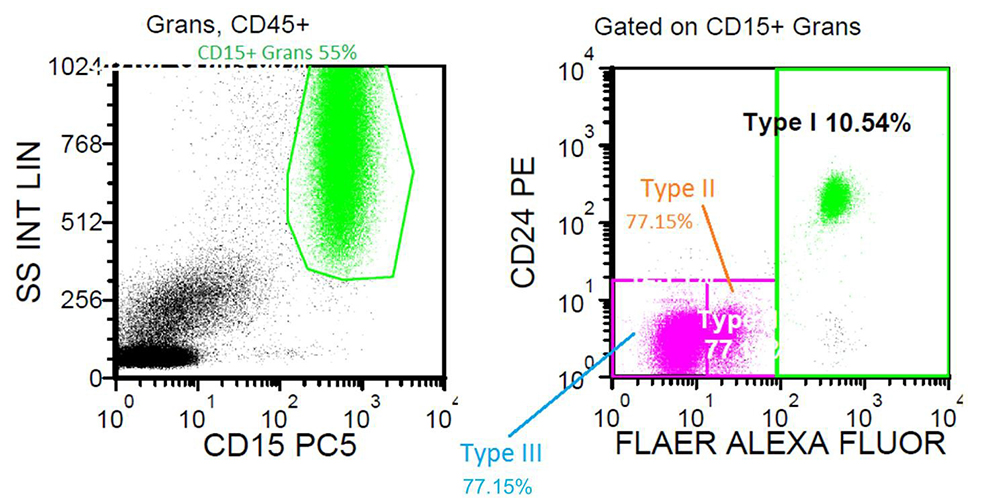
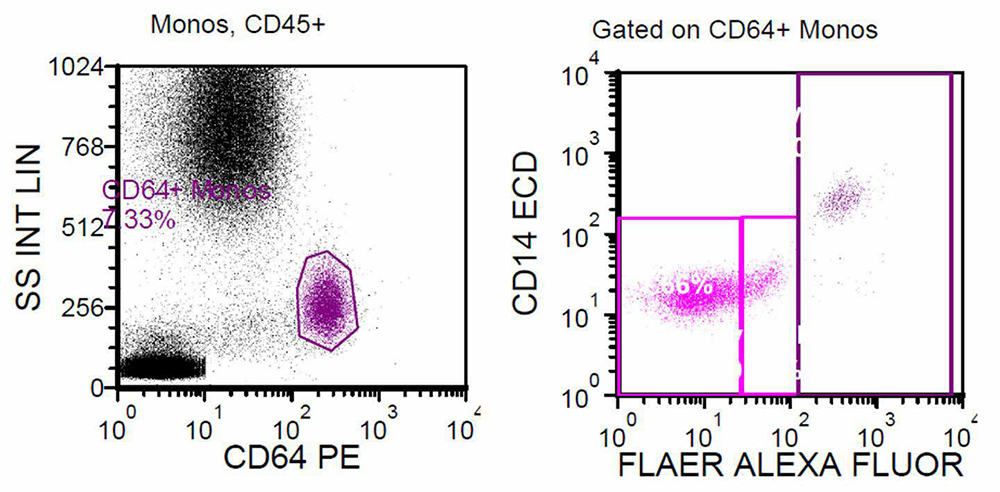
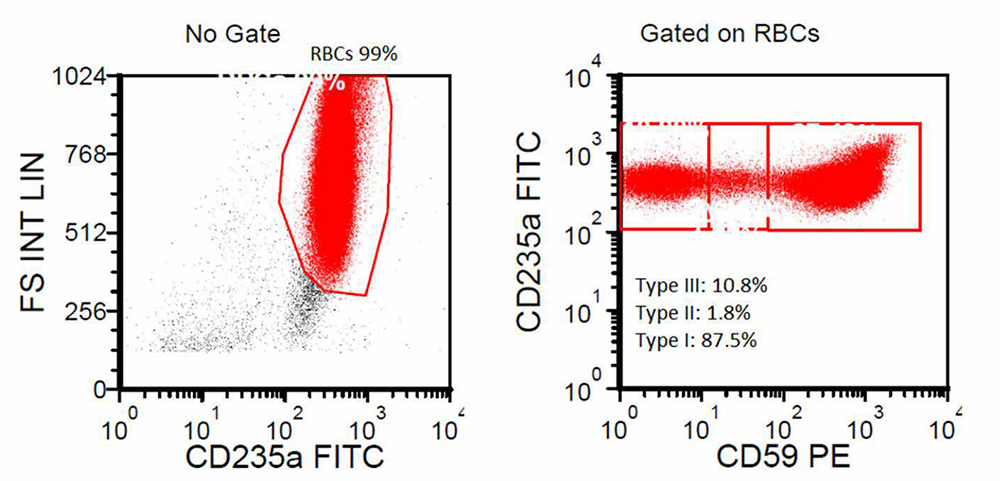
Diagnosis
Paroxysmal nocturnal hemoglobinuria
Discussion
Paroxysmal nocturnal hemoglobinuria (PNH) is an acquired hematopoietic stem cell disorder, producing abnormal erythrocytes, granulocytes, and platelets. PNH is caused by a sporadic mutation in the PIGA gene (phosphatidylinositol glycan anchor biosynthesis, class A) located on the X chromosome. To cause disease, the stem cell harboring the PIGA mutation must undergo clonal expansion, which occurs through an unknown mechanism. This mutation leads to reduced conjugation of certain proteins with the glycosyl phosphatidyl inositol (GPI) cell surface anchor. Associated GPI-anchored proteins include: CD55, CD59, C8-binding protein, CD58, CD14, CD24, CD16a, and CD157. The net result is decreased cell surface expression of these proteins, notably including CD59 (aka membrane inhibitor of lysis, or MIRL) and CD55 (decay accelerating factor, or DAF). Both of these proteins are involved in protecting the cell from complement-induced lysis, so the resultant loss of CD59 and CD55 on red cells makes them sensitive to lysis by the complement system, which causes subsequent anemia. A fluorochrome- conjugated derivative, fluorescent aerolysin (FLAER), has been developed such that it binds specifically to the GPI anchor, but does not lyse the cells. FLAER-based flow cytometric assays are more sensitive and accurate than assays based on individual antibodies to GPI-linked structures.
Patients with Coombs-negative hemolytic anemia, aplastic anemia, refractory anemia, and unexplained thrombosis in conjunction with cytopenias or hemolysis should be screened for PNH. However, it should be kept in mind that PNH clones could be seen in other disorders such as myelodysplastic syndrome. Since the FLAER testing is very sensitive, a small population of PNH cells could be detected by this assay. By using sensitive multiparametric flow cytometry, approximately 50–60% of patients with aplastic anemia and 15–20% of patients with low-risk MDS have been found to have a detectable population of GPI-AP-deficient erythrocytes and granulocytes.
References
- Hsi, Eric D., MD. Hematopathology, 3rd edition. Philadelphia, PA: Elsevier Inc; 2018
- McPherson, Richard A., MD, MSc, Pincus, Matthew R., MD, PhD. Henry's Clinical Diagnosis and Management by Laboratory Methods, 23rd edition. St. Louis, MO: Elsevier Inc; 2017
- Borowitz MJ, Craig FE, Digiuseppe JA, et al. Guidelines for the diagnosis and monitoring of paroxysmal nocturnal hemoglobinuria and related disorders by flow cytometry. Cytometry B Clin Cytom. 2010 Jul;78(4):211-30.
- Parker CJ. Paroxysmal nocturnal hemoglobinuria. Curr Opin Hematol. 2012 May;19(3):141-8.
Have Questions or Need Help?
If you have questions or would like to learn more about the Anatomic and Clinical Pathology Residency Program at Cedars-Sinai, please call or send a message to Academic Program Coordinator, LeeTanya Marion-Murray.
Department of Pathology and Laboratory Medicine
8700 Beverly Blvd., Room 8709
Los Angeles, CA 90048-1804