Research Areas
Pediatric Neurogenetic Disorders: Diagnosis, Disease Modeling and Therapeutic Interventions
Biomedical research often focuses on common disorders that affect a significant portion of the population (e.g., diabetes, cancer, heart disease). This approach prudently allocates limited research funds toward disorders that have the most widespread effect on collective medical needs. Unfortunately, this path often neglects rare disorders that affect only a handful of patients; however, the study of these rare disorders can be very valuable in providing insight into cellular and molecular functions.
Rare neurogenetic disorders are often caused by mutations that alter the function of important genes involved in neurodevelopment or function of cells in the nervous system. Many of the genes involved in neurodevelopmental disorders are crucial to cognitive development and human behavior; alternatively, pediatric neurodegenerative disorders occur after normal neurodevelopment with subsequent toxicity occurring due to lack of genes to maintain a functioning nervous system. The Pierson Laboratory studies both of these types of disorders with patient-derived IPSCs, NPCs, inducible-neurons and cerebral organoids to provide insight into neurologic function, as well as researches therapeutic interventions for affected families.
The recent emergence of genomic methods such as high-density single nucleotide polymorphism arrays, exome sequencing and whole-genome sequencing has revolutionized the ability to make diagnoses of rare or new disorders. Further advances in biotechnology, such as induced pluripotent stem cells, enables researchers to model these disorders with patient-derived cells to confirm the genetic diagnosis and research the mechanisms of disease. This knowledge sets up our ability to generate potential therapeutic interventions for these rare disorders.
We are thankful for the continued and generous support of the Cedars-Sinai Fashion Industries Guild for all of this work.
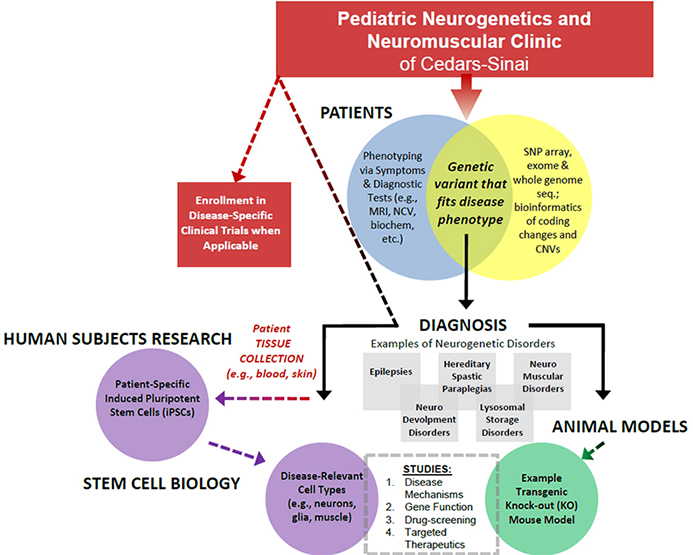
Schematic overview of the Pierson Lab’s pediatric neurogenetic disorder research. The Pierson Lab works with the Cedars-Sinai Pediatric Neurogenetic and Neuromuscular Clinic. The lab uses modern genomic platforms and induced pluripotent stem cell technologies to achieve genetic diagnoses and create cell culture models of these rare disorders, respectively.
Neurodevelopmental Disorders
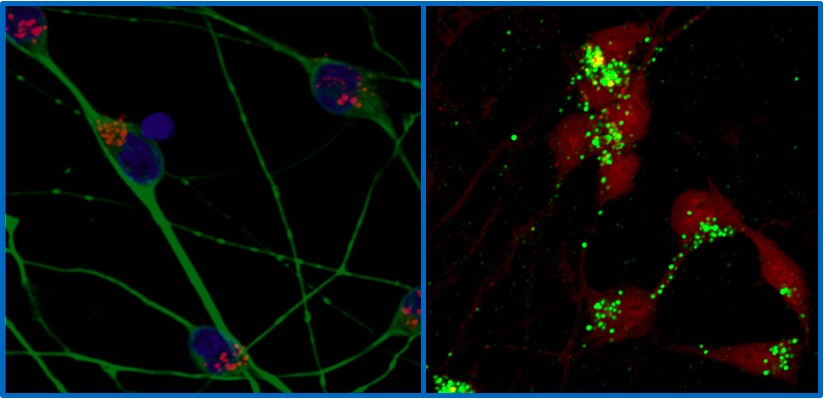
The development of induced pluripotent stem cells (IPSCs) has provided researchers with the ability to generate patient-derived stem cells that are capable of being differentiated into neural cells that include neurons, astrocytes and oligodendroglia. Currently, the Pierson Laboratory is modeling several neurodevelopmental disorders associated with intellectual disability, motor delays and apraxia of speech. For example, GATAD2B-associated neurodevelopmental disorder (GAND) is associated with mutations in the GATAD2B gene. GATAD2B encodes a protein that is involved in the NuRD complex, a multiprotein complex that regulates expression of key genes involved in neural differentiation and brain development. The Pierson Lab’s ongoing clinical study determining the genetic and clinical features of GAND is enrolling children with GATAD2B variants (contact tyler.pierson@cshs.org) to determine consistent and less frequent features of the disorder, while our laboratory is working with patient-derived GAND-IPSCs to evaluate target gene and protein expression in GAND-IPSCs, GAND-neuroprogenitors and GAND-neurons. As the NuRD complex plays a key role in cortical neuron differentiation, the Pierson Laboratory is evaluating RNA-seq data and cerebral organoid modeling to determine whether GAND causes incoordination of signals required for the proper cortical development. The lab is studying other neurodevelopmental genes (SATB2, CTIP2, or ARX) with these methods.
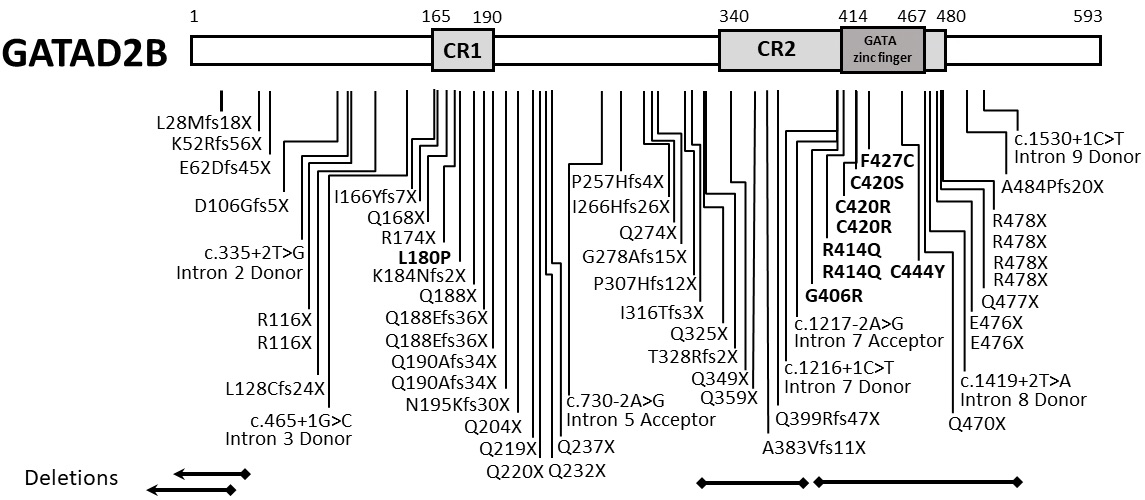
From Shieh C, … Pierson TM. GATAD2B-associated neurodevelopmental disorder (GAND): clinical and molecular insights into a NuRD-related disorder. Genet Med. 2020;22:878–888.
Pediatric Neurodegenerative Disorders
Another focus of the Pierson Laboratory and clinic is the study of pediatric neurodegenerative disorders. Lysosomal storage disease and mitochondrial disorders are areas of interest. For example, one of the Pierson Lab projects involves studying the molecular and cellular mechanisms behind a group of disorders called the neuronal ceroid lipofuscinoses (NCL). These disorders are characterized by a progressive clinical course in children that includes seizures, blindness and dementia as a result of the intracellular accumulation of storage material. CLN6 is one example of an NCL that is the result of mutations in the CLN6 gene encoding a resident endoplasmic reticulum transmembrane protein (CLN6p) whose dysfunction disrupts the autophagic-lysosomal system, and results in the accumulation of storage material leading to neuronal dysfunction and cell loss. The goal of the Pierson Lab is to generate cellular models of the CLN disorders to recapitulate the cellular phenotype in vitro. To accomplish this, the lab has differentiated CLN6-iPSCs with conventional and directed differentiation pathways including the inducible i3N system (Wang, et al. 2017). The i3N system allows for generation of TUJ1+ cells within three days of induction with DOX. The Pierson Laboratory hopes to produce CLN6 patient-derived iPSC models that can recapitulate CLN6 pathology in order to determine the pathologic mechanisms of the disorder, as well as target them with novel therapeutic screening pathways and interventions.